

Хромозомске патологије су један од најозбиљнијих поремећаја у ћелијама, због удвостручења једног од парова хромозома, настају посебни синдроми који имају типичне спољашње манифестације и промене у метаболизму. Тризомија у једном од парова хромозома (када уместо два постоје три хромозома) најчешће се налази у три варијанте - Довнов синдром, који је најчешћи и добро познат, као и Патау и Едвардс. Они су добили своје име због открића и описа од стране научника..
Општи подаци
Едвардсов синдром (или синдром трисомије 18) је на другом месту у смислу појаве трисомије, након Довновог синдрома, и јавља се када се 18. пар кромосома не разбије.. Патологију карактеришу различити поремећаји у формирању феталних система и органа у матерници, има типичне спољашње промене и неповољна здравствена и животна предвиђања..
Важно јеДјеца спадају у категорију особа са тешким инвалидитетом, захтијевају посебан третман, његу и бригу за своје родитеље и лијечнике..
 У погледу преваленције, ова хромозомска абнормалност се сматра врло честом, до 0,01-0,02% дјеце рођене у свијету има ову кромосомску абнормалност., и ниједна зависност од одређене расе или земље пребивалишта, је свеприсутна. Према статистикама, дјевојчице чешће трпе 3-4 пута чешће него дјечаци, иако за ову појаву нема добро утемељених знанствених података. Тачан разлог за настанак трисомије у фетусу није одређен, али је идентификован одређен број специфичних фактора ризика који повећавају вероватноћу његовог развоја..
У погледу преваленције, ова хромозомска абнормалност се сматра врло честом, до 0,01-0,02% дјеце рођене у свијету има ову кромосомску абнормалност., и ниједна зависност од одређене расе или земље пребивалишта, је свеприсутна. Према статистикама, дјевојчице чешће трпе 3-4 пута чешће него дјечаци, иако за ову појаву нема добро утемељених знанствених података. Тачан разлог за настанак трисомије у фетусу није одређен, али је идентификован одређен број специфичних фактора ризика који повећавају вероватноћу његовог развоја..
Уз друге хромозомске и генске поремећаје, данас Едвардсов синдром укључује конгениталне и неизљечиве патологије, посебне методе његе и медицинску подршку, рехабилитационе мјере омогућују да се на одређени начин подржи живот дјеце и да се оствари неки успјех у његовом развоју - физичком и менталном. Али због драматичне разноликости поремећаја које екстра хромозом даје, не постоје јединствене методе третмана и неге.
Историја проучавања патологије
У медицинским расправама и фикцији, у антици се могу наћи појединачне информације о пацијентима са овим синдромом, али је синдром детаљно описан почетком прошлог века. Развој технологија тог времена и раније није омогућио детаљно проучавање ове патологије, тако да разлози за то нису знали. Поред тога, већина дјеце рођене са овим синдромом умрла је у раној доби због лошег развоја медицине и благовремене помоћи њима. Узрок синдрома - трисомија на 18. пару хромозома откривен је тек 1960. године Д. Едвардсом, а по њему је названа нова патологија. Према медицинским статистикама, појава патологије у просјеку износи 1 случај на 3000 концепција, али постоји много мање фетуса прије рођења. То је због тога слична аномалија често на самом почетку завршава побачајом или смрћу фетуса у матерници. Али често, са таквим побачајима, нема хромозомских студија..
Шта је трисомија
Говорећи са становишта медицине, трисомија је варијанта хромозомских мутација, када пацијентове ћелије немају 46 хромозома склопљених у хомологне 23 парове, од којих један пар одређује полне разлике, али откривено је 47 хромозома..
Данас, лекари разликују три врсте трисомије које припадају несмртоносним мутацијама - Довн синдром (21 пар пати), Патау синдром (тринаести пар пати) и Едвардсов синдром (лезија је детектована на 18. пару). Све друге варијанте трисомије су у почетку неспојиве са животом, такви ембриони одмах умиру и трудноћа се не развија. Али рађање деце са ове три трисомије је увек проблем њиховог здравља, физичког развоја и интелигенције, менталних функција..
Узроци Едвардсовог синдрома
Као и друга два горе поменута патологија, Едвардсов синдром се назива генетским патологијама у којима се у аутосомима налази додатни хромозом (не сексуални хромозоми). Здрава особа има 46 хромозома - 22 пара - аутосоме (не-пол) и један пар - пол, утврђујући да ли је у питању девојка или дечак. ДНК ланци су компактно упаковани у хромозоме, у којима се бележе све генетичке информације о организму, почевши од процеса његовог зачећа до смрти. Цео програм живота је написан у ДНК ланцима..
Код Едвардсовог синдрома, тело не пати од дефеката гена, али хромозомски, постоји дефект у подели заметних ћелија, који затим даје живот детету, а додатни хромозом остаје у њему. У класичној верзији формирања Едвардсовог синдрома, долази до удвостручавања 18. хромозома, а и дечак и девојчица могу се родити са сличним синдромом.. У свим ћелијама таквог детета постоји вишак генетских информација. Оваква редундантност ДНК доводи до бројних поремећаја који доводе до спољашњих дефеката и проблема у раду система и органа. Због присуства трећег, додатног хромозома, појавило се научно име - трисомија.
Типови Едвардсовог синдрома: како зависи тежина манифестација
На основу тога какав је хромозомски дефект присутан у синдрому, постоје три врсте протока:
- Потпуна тризомија 18. пара.
- Делимична тризомија 18. пара
- Мозаички облик патологије.
Са фулл форм (такође припада класичном синдрому) претпоставља се да потпуно у свим ћелијама постоји додатни хромозом. Ова опција је најтежа и најчешће се дешава, прогнозе са њом су најнеповољније..
 Партиал трисоми односи се на ретке варијанте патологије, јавља се у 3% случајева, при чему овај проблем не садржи комплетан трећи хромозом, већ само његов комад. Овакав дефект може настати као посљедица проблема са дијељењем станица (посебно њихових кромосома), што је изузетно ријетко. Може бити и варијанта чињенице да је додатни део 18. хромозома везан за појединачне ДНК молекуле, што доводи до њиховог продужења и промене. У овим ћелијама постоје два 18. хромозома и део сувишног материјала. Тада конгенитални синдром неће бити толико озбиљан, јер се у ћелијама појављују додатне информације, не само о свим информацијама, већ ио њеним деловима.. Прогноза овог стања, иако неповољна, много је лакша за синдром него за пуни облик..
Партиал трисоми односи се на ретке варијанте патологије, јавља се у 3% случајева, при чему овај проблем не садржи комплетан трећи хромозом, већ само његов комад. Овакав дефект може настати као посљедица проблема са дијељењем станица (посебно њихових кромосома), што је изузетно ријетко. Може бити и варијанта чињенице да је додатни део 18. хромозома везан за појединачне ДНК молекуле, што доводи до њиховог продужења и промене. У овим ћелијама постоје два 18. хромозома и део сувишног материјала. Тада конгенитални синдром неће бити толико озбиљан, јер се у ћелијама појављују додатне информације, не само о свим информацијама, већ ио њеним деловима.. Прогноза овог стања, иако неповољна, много је лакша за синдром него за пуни облик..
Развој мозаички облик јавља се код 5-8% деце са сличном аномалијом. Али механизми овог типа синдрома су различити. Занимљиво је да се сличан облик развија након спајања сперматозоида са јајном ћелијом, а обе резултујуће ћелије у почетку имају нормалан сет хромозома - 46КСКС или 46КСИ. Али на самим првим поделама у образованој зиготи, настала је неисправност и једна од ћелија стечена овим додатним хромозомом. Као резултат тога, ембрион је добио ћелије са нормалним хромозомима - њих 46, други део има додатни хромозом - то јест, 47. Број ћелија са додатним хромозомом са таквим проблемима неће прећи 50%.
Обрати пажњуШто је већи број дефектних ћелија у ембриону и детету, то су лошија предвиђања, а њихов волумен зависи од времена када је дошло до неуспеха током поделе. Што се касније то десило, мањи је обим дефектних ћелија мрвица, а број развојних проблема је смањен..
Овај облик се назива мозаик јер су нормалне ћелије помешан са дефектним. Атипичне ћелије са додатним хромозомом су насумично разбацане по целом телу и нема начина да се уклоне на било који начин.. Овај облик је нешто лакши, трајање живота је дуже..
Основа патологије: шта је грешка гена?
Само један хромозом код дјетета има многе озбиљне и озбиљне посљедице по здравље и живот. То је због чињенице да у свим ћелијама постоји универзални систем за читање информација, и током дељења ћелија, удвостручење информација које су програмиране у ДНК молекулама. Ако постоји дефект најмање једног гена - он већ утјече на тијело у облику болести или проблема, и ако постоји цео комад екстра хромозома или чак читав хромозом, постоје вишеструке малформације и здравствени проблеми.
Хромозом 18 садржи до 2,5% свих генетичких информација о раду тела, а поремећаји који се репродукују због синдрома су веома значајни. Додатни гени на дефектном хромозому доводе до синтезе вишка протеина, што доводи до абнормалног развоја и функционисања више органа и система детета. Код Едвардсовог синдрома, скелет, одређене зоне нервног система, урогениталног система и кардиоваскуларног система су захваћене, имају и абнормалности у структури, а затим - како се фетус развија - у функционисању.
Према томе, главни и једини узрок овог синдрома је формирање додатног хромозома у полној ћелији мајке или оца. У производњи заметних ћелија не одвија се уобичајена подјела, након чега слиједи удвостручење 23 пара кромосома на 46 комада (за сваку идентичну копију у пару), већ само 23 комада. Ако у процесу дељења, 18. пар из неког разлога није правилно подељен, два хромозома одлазе у репродуктивну ћелију одједном, ау њој неће бити 23 комада, већ 24-д. Ако таква ћелија доводи до новог живота (без обзира да ли је то сперма оца или мајчино јаје), то ће довести до појаве детета са Едвардсовим синдромом..
Фактори ризика Едвардсовог синдрома
Да би се родитељске ћелије родитеља поделиле на погрешан начин, и оне које имају прекомерни сет хромозома, настају различити негативни утицаји, како споља тако и унутар тела.. Може се узети у обзир вођење:
- старост мајке и оца у време зачећа. Постоје реални докази о старосном фактору на вероватноћу трисомије. Како старимо, број абнормалних ћелија у репродуктивном систему се повећава, што је повезано са слабљењем контроле тијела над процесима поделе. Ризик од рађања деце са хромозомским патологијама после 30 година повећава се 2-3 пута у поређењу са младом годином, а након 40 година шансе се повећавају за 6-8 пута више. Зависност од очеве старости није тако очигледна, али су и хромозомски дефекти сасвим могући..
- имају лоше навике, посебно ако је у питању зависност од никотина и алкохола. Уобичајене интоксикације нарушавају процесе дељења ћелија, укључујући и гениталну област, што доводи до повећања ризика од неисправних спермија и јаја неколико пута. Још опаснија је употреба опојних дрога и токсикоманије, они даље нарушавају процесе диобе станица..
- узимам неке дроге, утиче на процесе дељења ћелија, вишка доза и трајање примене, неповољне комбинације лекова током третмана, формирајући њихове токсичне концентрације.
- соматске и гениталне инфекције, који доводе до пораза репродуктивних органа и ћелија, формирају хронични ток, утичу на процесе поделе (цитомегалија, херпес, папилома и др.).
- професионалне опасности, различите врсте изложености - јонизујуће, магнетне и многе друге. Они утичу на процес дељења ћелија, имају штетан утицај на ДНК, доводе до формирања слободних радикала у ћелијама и мутација.
Коначни узроци и сви фактори ризика који доводе до формирања ћелија са тризомијом могу се објаснити као открића молекуларне биологије и генетике. Сви ови фактори само повећавају ризик, али не кажите да ће се такво дијете родити..
Није склон формирању трисомије у заметним ћелијама, што доказује чињеница да се при рођењу у породици једног детета са Едвардсовим синдромом, шансе за рођење друге бебе повећавају 200 пута у поређењу са просеком - од 0,04% до 2-3% или више.
Карактеристике Едвардсовог синдрома у неонаталном периоду
Иако је данас могуће дијагностицирати синдром прије рођења, патологија се често јавља већ при рођењу. Новорођенче са сличном хромозомском аномалијом има типичне и изражене малформације, њихово присуство може помоћи у постављању дијагнозе чак и без додатних студија хромозома - генетичка анализа (али то се ионако ради, без ових података дијагноза није дефинитивна).
По рођењу, идентификовано:
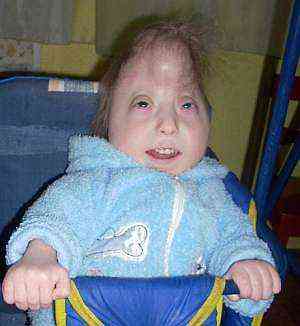 аномалије у структури облика лобање. Скелет са таквим синдромом је најизраженији - то се манифестује у промени првенствено у облику лобање са формирањем долихоцефалије (оштро издужена, уска лобања - "кула"). Таква аномалија није типична само за ову патологију, већ указује на постојеће развојне абнормалности. Најизраженија промјена у омјерима према мјерењима у подручју паријеталних костију и дужини од носног носа до окципиталних туберкула. Тежа и тежа аномалија лобање може бити микроцефалија, непропорционално мала величина лобање мозга у односу на тело..
аномалије у структури облика лобање. Скелет са таквим синдромом је најизраженији - то се манифестује у промени првенствено у облику лобање са формирањем долихоцефалије (оштро издужена, уска лобања - "кула"). Таква аномалија није типична само за ову патологију, већ указује на постојеће развојне абнормалности. Најизраженија промјена у омјерима према мјерењима у подручју паријеталних костију и дужини од носног носа до окципиталних туберкула. Тежа и тежа аномалија лобање може бити микроцефалија, непропорционално мала величина лобање мозга у односу на тело..- специфично модификован облик ушне шкољке. Абнормална структура ушију код новорођенчади је изразито изражена и примећена је код 95% беба, посебно са пуним типом синдрома. Ушне шкољке су много ниже него иначе, релативно оштро спуштене у односу на орбите, хрскавице су конвексне и немају јасно обликоване коврче. Ушна шкољка и трагус су у раном дјетињству или су потпуно одсутне, слушно месо нагло се сужава и четвртина дјеце може бити потпуно одсутна..
- абнормална структура неба са доњом вилицом (формирање микрогнатије). Код новорођенчади, када се сумња на Едвардсов синдром, нема потпуног нагомилавања непчаних процеса у горњој чељусти, који чине уздужни прорез. Може постојати опција са тоталном дисасоцијацијом само меког непца или парцијалне фузије, понекад са преласком на усне. Са потпуним обликом дефекта, не-дисјунктност може бити билатерална, горња усна је подељена са обе стране, што доводи до патолошких пукотина у вилицама и непцу, што спречава дете да потпуно затвори уста. У случају тешких патологија, могућа је комуникација између орофаринкса и носне шупљине, чак и ако су уста затворена. Учесталост таквих аномалија достиже 20-25% и више..
- Могу постојати и други проблеми. - готхиц ски (претјерано високи свод костију) и промене у структури мандибуле. Они су регистровани у око 70% деце. Типично мицрогнатхиа - брада и доња вилица су снажно повучене, што спречава бебе да потпуно затварају уста и цурење пљувачке из уста. Са таквом аномалијом постоје потешкоће у исхрани, посебно наношењем на дојку..
- деформације стопала са формирањем "ножног љуљања", карактеристичне аномалије дужине прстију екстремитета. Промјена стопала је типична за 75% или више дјеце на позадини овог синдрома, долази до промјене положаја костију - глежња, пета или скафоида, што доводи до формирања снажно истакнуте пете против одсуства лука стопала. Може се савити према напријед, дајући ногама изглед ногу од столице за љуљање. Такође се откривају неравнотеже у дужини прстију ногу, посебно великих. Краћи је од другог ножног прста на нози, као што се види када су прсти спљоштени.
- руке у флексор положају и адхезије између прстију, специјални дерматоглифски цртежи. Синдацтили (овај термин се односи на фузију прстију) детектује се у 45% случајева, обично је то прст, али руке могу бити погођене. У благом степену, кожа ствара мембране, а коштани надвоји могу настати у тешким. Може бити и флексорске аномалије руку - положај прстију у аномалном, стално затегнутом положају, прекривањем малог прста и палца свих осталих, чврсто притиснутих на длан. Такви поремећаји су типични за 90% дјеце са синдромом..
- Између осталог, са карактеристичном трисомијом шаре на прстима и длановима са посебним обликом кожних набора. Са развојем овог синдрома, абнормалности су типичне за 60% беба. То су врло чести лукови на јастучићима, одсуство кожног набора између екстремних и претпоследњих фаланга, развој трансверзалног жлеба - има израз "мајмунска линија"..
- гениталне малформације. Код дечака то је неразвијеност пениса и скротума, код девојчица, повећање величине клиториса. Аномалије се обично комбинују са дефектима генитоуринарног система, али очигледно да остали знаци не виде.
 аномалије у структури облика лобање. Скелет са таквим синдромом је најизраженији - то се манифестује у промени првенствено у облику лобање са формирањем долихоцефалије (оштро издужена, уска лобања - "кула"). Таква аномалија није типична само за ову патологију, већ указује на постојеће развојне абнормалности. Најизраженија промјена у омјерима према мјерењима у подручју паријеталних костију и дужини од носног носа до окципиталних туберкула. Тежа и тежа аномалија лобање може бити микроцефалија, непропорционално мала величина лобање мозга у односу на тело..
аномалије у структури облика лобање. Скелет са таквим синдромом је најизраженији - то се манифестује у промени првенствено у облику лобање са формирањем долихоцефалије (оштро издужена, уска лобања - "кула"). Таква аномалија није типична само за ову патологију, већ указује на постојеће развојне абнормалности. Најизраженија промјена у омјерима према мјерењима у подручју паријеталних костију и дужини од носног носа до окципиталних туберкула. Тежа и тежа аномалија лобање може бити микроцефалија, непропорционално мала величина лобање мозга у односу на тело..Поред ових манифестација, јављају се и мањи знаци и аномалије у развоју деце, који могу у прелиминарној дијагнози синдрома. Важно је напоменути да један од наведених симптома, па чак и два још увијек не говоре о дијагнози, постоје вишеструки дефекти у синдрому и имају различите комбинације..
Важно јеВећина описаних манифестација може се појавити као појединачне варијанте и не потврђују дијагнозу, могу бити изолована варијанта абнормалности током трудноће или манифестација других патологија. Само дијагноза вањских дефеката не може се направити.!
Штавише, на основу непотпуних или мозаичких типова синдрома спољних знакова, може бити и један или два, док постоје озбиљни дефекти унутрашњих органа са честим абнормалностима у хромозомима. Они одређују будуће прогнозе за живот и ток синдрома..
Развој детета: карактеристике Едвардсовог синдрома
Само појава са појавом светлости и манифестације малформација у развоју, Едвардсова аномалија није ограничена, како расте и развија се, могуће су манифестације нових и тежих одступања. Први од њих се може идентификовати у року од неколико недеља од тренутка рођења. Често, по први пут, могуће је посумњати на дијагнозу мозаика и непотпуне врсте патологије, ако постоји мало или никаквих вањских дефеката.. Горе описани дефекти и спољашње промене постају оштрије и израженије како расту, постепено се додају нови проблеми и аномалије које нису приметне при рођењу. Дакле, овде вреди укључити:
- Оштро и изражено заостајање у нивоу физичког развоја. При рођењу маса такве дјеце не прелази 2-2,5 кг, а разлог за то су кромосомски дефекти, који не дозвољавају да се системи и органи у потпуности развију. Како развој напредује, раст и телесна тежина значајно варирају од мјесеца до мјесеца, а опсег груди је такођер слаб. Величина главе може да достигне или изнад нормале, што може указивати на акумулацију вишка течности унутар кранијалне шупљине, формирајући хидроцефалус.
- Проблеми са стопалима. Како расту, формира се стопало услед аномалија у структури костију и лигамената, мишићног хипертонуса. Патња и контрола развоја стопала и тонуса нервног система. Ово је угрожено чињеницом да деца почињу да ходају касно или то уопште не могу да ураде због општег озбиљног стања. Споља, клекавица се манифестује у тешком деформитету стопала и његовој ненормалној инсталацији у мировању.
- Поремећај тонуса мишића. При рођењу, хипертоничност је типична за руке, формирајући њихову позицију флексора, али како се развија, шири се на сусједне групе мишића, доводећи до артистичких положаја. Међутим, чешће се смањује тонус мишића, деца су трома и атонична, екстремитети су мекани бичевима. Такође могу постојати стања дистоније са оштром контракцијом појединачних мишићних група у односу на позадину хипотензије других. У овом случају, руке могу бити савијене, а ноге се могу савити. То доводи до нарушене координације и хаотичних покрета, дислокација и абнормалног положаја удова..
- Атипичне реакције нервног система, емоционални проблеми. Неки делови мозга код деце са Едвардсовим синдромом су недовољно развијени, па се стога стварају проблеми емоционалности и инхибиције, неадекватне реакције на околину. Обично се повезује са цорпус цаллосум и церебеларним хипоплазијама, које такође формирају ментална ретардација. Вањски се то манифестује одсутним погледом, недостатком емоција и немогућношћу успостављања контакта, одсуством објеката за праћење. Деца не реагују на звукове због проблема са ушима и нервним системом, такве аномалије се откривају у првим месецима живота..
Генерално, због тешких и комбинованих лезија, деца не живе до пунолетства.. Неки од њих са мозаичном формом могу да досегну старост адолесцената са дубоким степеном имбецилности и спољашњим манифестацијама малформација..
Дијагностичке технике Едвардовог синдрома
Данас постоје три типа дијагностичког синдрома, који су фундаментално различити.. Имајући у виду чињеницу да је патологија неизлечива и смртоносна у раном узрасту, важно је обратити пажњу на пренаталну дијагностику, која не дозвољава рађање дјеце са овим абнормалностима.. Консултације генетичара у породицама у којима је било случајева хромозомских абнормалности такође ће бити од помоћи.. Можете потрошити:
- Дијагноза заметних ћелија пре зачећа. Нажалост, овај метод је још увек веома ограничен и само у ИВФ протоколима у прелиминарном истраживању заметних ћелија. Ако је ово природна концепција, доктори могу предвидјети веће ризике за рађање такве дјеце, али то није ништа друго до предвиђања. Немогуће је са сигурношћу знати да ли ће се такво дијете родити или не. Анализира се породична анамнеза узимајући у обзир све постојеће болести, процењују се фактори ризика и врши се генетска анализа за оба родитеља коришћењем софистициране опреме и тестова. Одређивањем кариотипа родитеља могу се извући одређени закључци..
- Идентификација синдрома током гестације. Са развојем фетуса, постоји неколико метода за директну или индиректну потврду Едвардсовог синдрома, у почетку се ултразвучни скрининг и тестови крви користе у ове сврхе уз проучавање специфичних индикатора, и код високог ризика, проводе се инвазивне процедуре - амниоцентеза, или цордоцентесис и биопсија хорионских вила. Проводити скрининг у строго дефинисаним периодима, када ће промена података о ултразвуку и крвних параметара бити најзначајнија.
Добијање директних ћелија фетуса или његове крви омогућава вам проучавање њених хромозома и потврђивање или одбијање Едвардсовог синдрома. Ово помаже у одлучивању о томе да ли ће продужити или прекинути трудноћу из медицинских разлога..
- Потврда синдрома након рођења. Спроводи се према резултатима екстерних података у комбинацији са низом генетских анализа које потврђују присуство трисомије на 18. пар хромозома. Поред тога, важно је процијенити стање дјетета и предвидјети његов живот и здравствено стање како би се схватило колико му је помоћи да преживи.. Без медицинске заштите за овај синдром, деца могу брзо да умру на пољу рођења у наредним недељама.. Ултрасонографија и ехокардиограм, радиографија и ЕЕГ, разне анализе крви и урина, додатни тестови до МРИ и ЦТ.
Третман и прогноза за Едвардсов синдром
Нажалост, овај синдром је до данас неизлечив, а лекари могу само помоћи дјетету да се боље носи са различитим проблемима кроз хируршке интервенције и конзервативну терапију.. Обично дјеца живе до неколико мјесеци или годину дана, само дјеломично или мозаично могу живјети дуже. Смрт долази од вишеструких малформација и абнормалности у раду органа, као и спајања повезаних компликација. Радикално исправљање дефеката хромозома у ћелијама дететовог тела у садашњој фази развоја медицине је немогуће.
Алиона Паретскаиа, педијатар, лекар