

Мукополисахаридоза је читав низ генетски одређених (условљених) патологија које се јављају у случају поремећаја метаболизма киселих мукополисахарида (гликозаминогликана)..
Најчешће, са таквим прекршајем, костур је захваћен и физички развој се одлаже, а неки типови патологије манифестују се назадношћу интелектуалне сфере. Поред тога, постоје кварови у кардиоваскуларном, гастроинтестиналном и нервном систему, оптичким апаратима и кожи..
Лечење мукополисахаридозе је симптоматско, прогноза је генерално неповољна - оштећења напредују јер се глукозаминогликани акумулирају. Пацијенти умиру довољно рано.
Општи подаци
Мукополисахаридоза је проучавана релативно недавно - у 20. веку. У почетку се сматрала јединственом патологијом, па су због различитости почели да разликују њене типове. Уједињујући фактор све мукополисахаридозе је да се киселински мукополисахариди акумулирају у органима и ткивима, а узрок ове повреде је инфериорност неких ћелијских ензима, који је инкорпориран у генотип и наслеђен је.
Обрати пажњуОртопеди лече болеснике са мукопополисахаридозом, али захтевају и клиничке напоре кардиолога, офталмолога, неуролога, оториноларинголога и неких других специјалиста..
Мукопополисахаридоза типа ИХ
Мукополисахаридоза типа ИХ се такође назива Хурлеров синдром. Дијагностикован је код 1 новорођенчета за 20-25 хиљада.
Први знаци болести почињу да се јављају током прве године живота детета, али се комплетна клиничка слика може развити само за 2 године..
Најчешћи симптоми болести су:
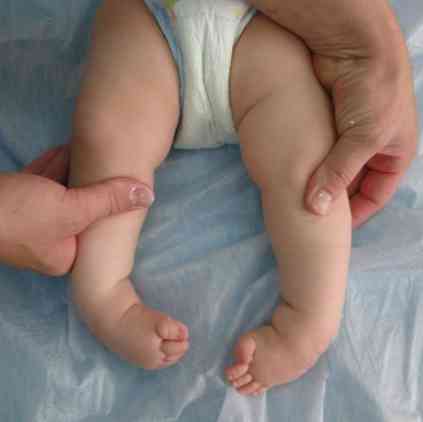 грубе обрисе лица - врло типично за ову патологију;
грубе обрисе лица - врло типично за ову патологију;- деформација лобање - постаје као кобилица брода. Ово стање се назива сцарацепхалиа;
- дисање се изводи кроз уста, јер је дисање у носу смањено због повећања аденоида, као и интраутериних развојних абнормалности у лобањи лица;
- деформације екстремитета и неких других фрагмената костура;
- успоравање раста;
- закривљеност кичме (симптом "мачка леђа" у седећем положају);
- скраћивање врата;
- атипични високи распоред лопатица;
- испупчена предња доња ребра;
- повећање ширине четкица - док наликују канџи са канџом;
- развој контрактура (смањење опсега покрета) уз постепено повлачење различитих зглобова у патолошки процес. Прво су захваћени зглобови лакта и рамена, а процес се протеже до зглобова колена, кука и скочног зглоба.
 грубе обрисе лица - врло типично за ову патологију;
грубе обрисе лица - врло типично за ову патологију;Хурлеров синдром карактерише специфичан ход - на прстима са савијеним ногама. Таква карактеристика настаје због ограничења покрета због контрактура..
Физички преглед открива следеће:
- трбух је увећан, предњи зид код таквих пацијената ослабљен, па се често могу развити умбиларне и ингвиналне киле;
- увећана јетра и слезина;
- код мушкараца се често открива хидрокела - воденица тестиса;
- у неким случајевима се откривају повреде координације покрета;
- открила је прекомерни раст длакаве косе.
Када се инструментални преглед утврђује на следећи начин:
- на електрокардиограму - знакови дифузног (распрострањеног) оштећења срчаног мишића;
- оштећење вида и слуха;
- Офталмоскопија открива замућење и повећање рожњаче.
Најкарактеристичније су промене које се јављају током рендгенског прегледа:
- Рентген кичме - омогућава вам да идентификујете карактеристичну деформацију кичменог стуба. У овом случају, пршљенови имају облик коцке, а њихове ивице су заобљене. Такође обележена кифоза - закривљеност кичме напред;
- рендгенски снимак грудног коша - наглашено задебљање предњих фрагмената ребра са истовременим стањивањем њихових постериорних делова, скраћивањем и деформацијом. Ребра истовремено добијају специфичан изглед и постају слична лопати или сабљи. Забележено је смањење и померање глава хумеруса;
- радиографија карлице - открива сужавање карличног прстена, ацетабуларне шупљине (оне у којима је "основа" формирања зглоба кука) су искривљене, а главе фемура смањене;
- Уочен је рендгенски снимак тубуларних костију - стањивање кортикалног (површинског) слоја костију и извјесна експанзија медуларног канала;
- Рендгенски снимак костију руке - помаже у откривању неразвијености дисталних (ноктних) фаланга прстију, скраћивања и ширења средњих и проксималних (ближе длановима) фаланга, као и метакарпалних костију;
- Рендгенска слика лобање - јасно неразвијене кости лобање лица, краниостеноза (сужавање лобање на неким локацијама) и макроцефалија (повећање лобање).
Код пацијената са дијагнозом мукополисахаридозе типа ИХ идентификовани су следећи поремећаји, који се могу сматрати посебним болестима:
- ретинална пигментна дистрофија - кршење њеног развоја;
- глауком - повећање интраокуларног притиска;
- конгестија у фундусу;
- пареза - поремећај кретања;
- парализа - потпуно прекидање моторичке активности у одређеним подручјима мишићно-скелетног система.
Временом, пацијенти са дијагнозом Хурлера развијају и неповратно напредују у менталној ретардацији.
Мукопополисахаридоза типа И-С
Још једно име за мукополисахаридозу типа И-С је болест болести. Болест је описана тек средином 20. века, иако су пацијенти са карактеристичним контрактурама прстију руку, поремећајима кретања у великим зглобовима и специфичним особинама лица раније били присутни у пракси клиничара. Симптоми се могу комбиновати са знаковима Хурлеровог синдрома.
Овај тип мукополисахаридозе се јавља у каснијим годинама него код мукополисахаридозе типа ИХ, а његов ток је повољнији..
Карактеристична особина мукопополисахаридозе типа И-С је да до 3-6 година старости развој дјеце одговара старосној норми. Даље, почињу да се појављују следећи знаци:
- флексијске контракције прстију;
- ограничење проширења у великим и средњим зглобовима - наиме, лакат, рамена и зглоб.
Повреда амплитуде покрета на доњим екстремитетима је мање изражена него код других типова описане патологије.
Потпуна клиничка слика овог типа мукополисахаридозе утврђена је почетком адолесценције.
Физиолошка студија (испитивање) наводи следеће:
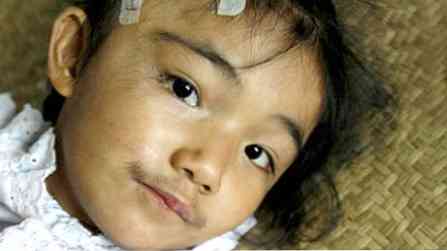 пацијенти нису високи, али имају јаку грађу (здепаст);
пацијенти нису високи, али имају јаку грађу (здепаст);- карактеристике су прилично грубе;
- добро развијена скелетна мускулатура;
- примећена је хипертрихоза (повећана длака на телу);
- кожа на прстима руку (чешће) и стопала (рјеђе) је згуснута и растегнута;
- величина јетре и слезине, по правилу, није промењена.
 пацијенти нису високи, али имају јаку грађу (здепаст);
пацијенти нису високи, али имају јаку грађу (здепаст);Поред тога, код ових пацијената се идентификују повреде које се могу сматрати посебном болешћу:
- ингвинална или умбиликална хернија;
- синдром карпалног тунела - развија се услед наглашене компресије средњег нерва. Она се манифестује атрофијом (хипоплазијом) тенарских мишића (елевација у подручју палца) и парестезијама (поремећај осјетљивости) у подручју 3, 4 и 5 прстију;
- аортна стеноза - сужавање аорте;
- инсуфицијенција аорте;
- ретинална пигментна дистрофија;
- глауком;
- замрачење рожњаче.
Интелигенција пацијената са дијагнозом мукопополисахаридозе типа И-С остаје нормална, за разлику од пацијената са мукополисахаридозом типа ИХ.
Од инструменталних метода најинформативнија је радиографија. Идентифицира промјене које су сличне промјенама у мукополисахаридози типа ИХ, али су мање изражене..
Мукополисахаридоза типа ИИ
Овај тип мукополисахаридозе се назива и Хунтерова болест. Патологија најчешће погађа дечаке од 2-3 године.
Према клиничким манифестацијама, болест је слична мукополисахаридози типа ИХ - док постоје:
- сцариоцепхали;
- грубе карактеристике;
- низак тон гласа;
- отежано дисање - због деформације костију скелета лица.
Са даљим напредовањем болести придружују се следећи знаци:
- смањена интелигенција;
- координација покрета је прекинута;
- Расположење у таквом пацијенту је нестабилно - уочава се такозвана емоционална лабилност - од напада смеха до плача. Такве промјене расположења могу се појавити у било које вријеме..
Са прогресијом мукопополисахаридозе типа ИИ појављује се и повећава неоправдана агресивност.
За разлику од мукопополисахаридозе типа ИХ, повреде кичме у облику кифозе нису карактеристичне, симптом "мачје леђа" није уочен.
Физички преглед одређује следеће:
- откривено је благо повећање слезене и јетре;
- на кожи леђа су одређени специфични нодули.
Када је инструментални преглед открио:
- непрозирност рожнице, која напредује;
- повећање губитка слуха.
Од свих инструменталних метода дијагностике, најинформативнија је радиографија разних фрагмената мишићноскелетног система - лобање, рудна ћелија, карлица и тако даље. У исто време, радиолошки знаци су исти као и за мукополисахаридозу типа И-С.
Пацијенти са таквом дијагнозом су склони развоју болести респираторног система:
- трацхеитис;
- бронхитис;
- пнеумонија.
Треба напоменути да се мукопополисахаридоза типа ИИ може развити у облику две опције:
- неповољан (опција А);
- повољно (опција Б).
 Неповољну варијанту показује јасна клиничка слика, при чему се уочавају битни поремећаји интелигенције. До смртних случајева може доћи током адолесценције.
Неповољну варијанту показује јасна клиничка слика, при чему се уочавају битни поремећаји интелигенције. До смртних случајева може доћи током адолесценције.
Симптоми са повољном варијантом нису изражени, а горе наведена кршења могу бити праћена и благом менталном ретардацијом. Такви пацијенти, по правилу, живе до 30 година, у неким случајевима - и више.
Мукополисахаридоза типа ИИИ
Мукополисахаридозу типа ИИИ описао је амерички педијатар Санфиллипо, тако да је патологија добила друго име - Санфиллипо болест.
Болест се дијагностикује код 1 дјетета на 100-200 тисућа новорођенчади..
Деца са типом мукополисахаридозе типа ИИИ у првим годинама живота развијају се у складу са старосним нормама, а могу се приметити и неке манифестације патологије - нарочито тешкоће у гутању и неспретан ход..
Почевши од 3-5 година, дете почиње да заостаје за својим вршњацима у физичком развоју, а истовремено постаје апатично, равнодушно према људима и догађајима.. Типични знаци су:
- оштећење говора;
- храпавост карактеристика;
- редовна уринарна инконтиненција (не само ноћна) и измет.
Како дете расте, ментална ретардација се повећава и на крају постаје главни симптом овог типа мукополисахаридозе..
Физички преглед је открио:
- незнатно повећање јетре и слезине;
- равномерно повећање дистрибуције косе;
- контрактура;
- успоравање раста.
 Кршења органа вида и кардиоваскуларног система, који се често јављају у другим облицима описане патологије, за овај тип мукополисахаридозе нису типични.
Кршења органа вида и кардиоваскуларног система, који се често јављају у другим облицима описане патологије, за овај тип мукополисахаридозе нису типични.
Резултати рендгенског прегледа са описаном патологијом или без патолошких промена, или слични онима који су детектовани са мукополисахаридозом типа И-С, али мање израженим \ т.
Прогноза за ову болест је мање повољна него за друге типове мукополисахарозе - такви пацијенти умиру у доби од 10 до 20 година, инфективне компликације постају узрок смрти..
Мукопополисахаридоза типа ИВ
Мукополисахаридоза типа ИВ се назива и Моркио болест (названа по уругвајском педијатру који ју је описао). Патологија је чешћа у односу на друге типове мукополисахаридозе - наиме, код 1 детета на 40 хиљада новорођенчади.
Деца са овом болешћу до 1-3 године се развијају без особина. Али онда се појављују следећа кршења:
- дете значајно заостаје за својим вршњацима;
- у процесу развоја уочио је скраћивање врата и торза;
- развијају се разне контрактуре;
- кожа се згусне;
- смањује се снага мишића;
- слух се погоршава;
- црте лица постају грубе;
- јављају се разне деформације груди.
Такође за мукополисахаридозу типа ИВ постоје повреде које се могу сматрати засебним патологијама:
- валгусна деформација удова - закривљеност у вањском смјеру;
- сколиоза (закривљеност кичме у латералној равни) или кифоза;
- ингвинална кила;
- умбиал херниа;
- дистрофија рожнице.
Интелект са овим обликом мукопополисахаридозе је сачуван.
Дијагноза описане патологије потврђује се рендгенским снимањем, у овом случају је најинформативнија. То су следећи типови:
- Рендгенски преглед кичменог стуба - слике показују закривљеност кичменог стуба у облику кифозе и сколиозе, као и експанзију и спљоштеност краљешака;
- радиографија карлице - откривене вишеструке деформације прстена здјеличне кости, неједнакост његових контура;
- Рендгенски снимци удова - изравнавање главе бутне кости, експресивно скраћивање костију подлактице и откривање деформитета костију стопала.
Пацијенти са мукопополисахаридозом типа ИВ живе, по правилу, мање од 20 година. Фатални исход се дешава на позадини низа повезаних болести, које су на крају компликоване кардиопулмоналном инсуфицијенцијом..
Мукопополисахаридоза типа ВИ
Још једно име за мукопополисахаридозу типа ВИ је Марото-Ламијева болест (након имена два француска доктора који су описали ову патологију)..
Клинички знаци су углавном "одјекивали" са симптомима других типова мукополисахаридозе, али најизраженији су:
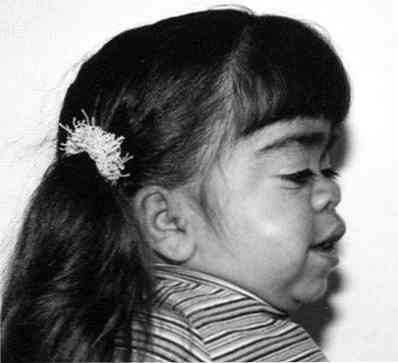 значајна грубост црта лица;
значајна грубост црта лица;- приметно заостајање у расту;
- скраћивање врата - понекад се чини да пацијентова глава "седи" готово на рамену;
- деформитет грудног коша.
 значајна грубост црта лица;
значајна грубост црта лица;Такви пацијенти врло често имају прехладе - овај симптом омогућава да се посумња на овај, а не други облик мукополисахаридозе..
Физички преглед открива следеће:
- јетра и слезина су често увећане;
- може се дијагностицирати ингвинална кила.
Интелектуално, дијете се развија без кршења - има способност размишљања и рјешавања интелектуалних проблема..
Постоје две варијанте овог типа мукополисахаридозе:
- цлассиц;
- мукополисахаридоза са мањим клиничким манифестацијама.
Да бисте потврдили дијагнозу, помоћи ћете рендгенском прегледу - слике се одређују према:
- деформација пршљенова - они постају коцкасти или клинасти;
- троугласта деформација карличног прстена;
- хипоплазија (неразвијеност) и деформација главе бутне кости;
- скраћивање фибуле.
Мукопополисахаридозе типа ВИИ и ВИИИ
 Мукополисахаридоза ових типова је рјеђа у односу на друге типове описане патологије..
Мукополисахаридоза ових типова је рјеђа у односу на друге типове описане патологије..
Мукополисахаридоза типа ВИИ клинички се јавља као тип ИИИ болести, разлика између њих се налази само у резултатима биохемијских студија..
Мукополисахаридоза типа ВИИИ је симптоматска и слична мукополисахаридози типа ИВ. Али постоји разлика у манифестацијама - то је повреда интелектуалног развоја, која се не примећује код типа ИВ мукопополисахаридоза.
Дијагностика
Приликом постављања дијагнозе мукополисахаридозе, оне се заснивају на притужбама, анамнестичким подацима, резултатима додатних метода испитивања - физичком, инструменталном, лабораторијском. Може се посумњати на присуство мукополисахаридозе по карактеристичним деформитетима костура, који "захваћају око", али је могуће одредити тип описане болести само уз помоћ додатних метода испитивања.. Главне методе су:
- радиографија;
- откривање гликозаминогликана у урину;
- одређивање ензимске активности у ћелијским културама.
Код клиничких знакова поремећаја унутрашњих органа укључени су начини испитивања:
- електрокардиографија;
- ултразвук јетре и слезине;
- електроенцефалографија;
- биохемијски тест крви
и многе друге.
Диференцијална дијагностика
Диференцијална (препознатљива) дијагноза се изводи између различитих типова мукополисахаридозе.
Компликације
Најчешће компликације мукополисахаридозе су:
- контрактура;
- затајење срца;
- плућна инсуфицијенција.
Третман
Није развијена терапија за мукополисахаридозу, која се може користити за елиминисање болести, а не само њених манифестација.. Такви пацијенти примају симптоматско лечење - то може бити:
- конзервативна - трансфузија крви, хормонска терапија, витаминска терапија и тако даље;
- оперативна - ендопротеза глава бутне кости, поправка киле, хируршка интервенција за отклањање контрактура и исправљање деформитета скелета и других.
Важни су:
- лечење инфекција респираторног тракта;
- корекција вида и оштећења слуха.
Превенција
Пошто се патологија јавља због генетских поремећаја, не постоје специфичне методе превенције.. Међутим, ризик од поремећаја гена који може довести до развоја различитих типова мукополисахаридозе може се смањити ако се женама пруже нормални услови трудноће. Ово је:
- одбацивање лоших навика;
- придржавање рада, одмора, сна;
- оптимистично расположење.
Форецаст

Прогноза за мукополисахаридозу је генерално неповољна, то се односи на све типове описане болести. У процесу живота и развоја у ткивима прогресивно се акумулирају метаболички продукти који настају због недостатка ткивних ензима. То доводи до патолошких промена од стране готово свих органа и система. Ако је патологија започета, смрт може настати не само због кардиопулмонарног, већ и полисистемског неуспјеха.
Конзервативна терапија игра улогу подршке - употреба било којег лијека доводи до привременог побољшања, али не може надокнадити недостатак ткивних ензима.
Ковтониук Оксана Владимировна, медицински коментатор, хирург, консултантски лекар